AGENNIX


-
CAR-T & CAR-NK Cell Therapies
CAR-T & CAR-NK Cell Therapies Cellular immunotherapy has emerged as a groundbreaking approach in cancer treatment, with Chimeric Antigen Receptor (CAR) engineered T and Natural Killer (NK) cells at... -
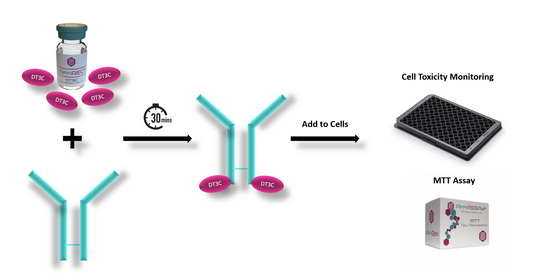
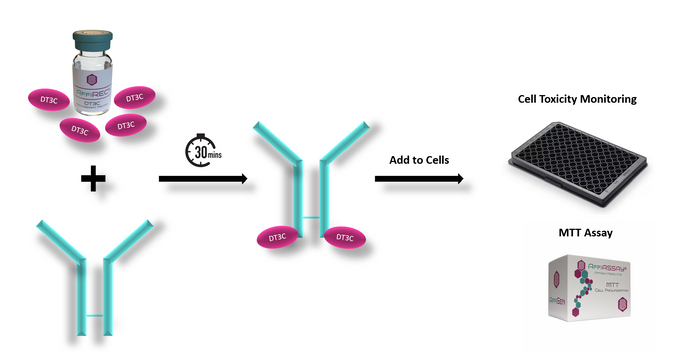
ADCs Development: Our Solutions for Evaluation of Internalization Efficiency in Cancer Therapy
ADCs Development: Our Solutions for Evaluation of Internalization Efficiency in Cancer Therapy ADCs Development: Our Solutions for Evaluation of Internalization Efficiency in Cancer Therapy ADCs Development: Our... -
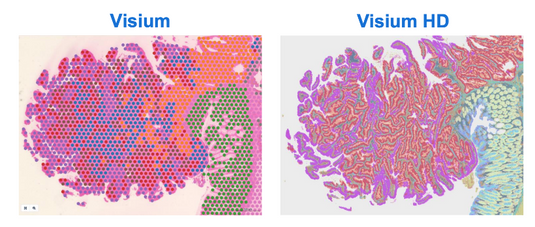

Visium HD Spatial Gene Expression: Revolutionizing Biological Insights
In the rapidly evolving field of genomics, the ability to map the gene expression of tissues with high resolution is paramount. Visium HD Spatial Gene Expression technology represents a... -
The Challenge of Genomic DNA Contamination in cDNA Synthesis: Strategies and Solutions
The Challenge of Genomic DNA Contamination in cDNA Synthesis: Strategies and Solutions In the realm of molecular biology, unraveling the intricacies of gene expression is paramount to understanding cellular... -
cDNA Synthesis using AffiPCR® All in One RT-PCR Master Mix with gDNA Removal
cDNA Synthesis using AffiPCR® All in One RT-PCR Master Mix with gDNA Removal In the dynamic realm of molecular biology, the quest for streamlined and precise methodologies is incessant. As... -


AffiELISA® CLIC1, CLIC2, CLIC3, CLIC4, CLIC5, and CLIC6 ELISA Kits
Overview Chloride intracellular channel proteins (CLICs) are a unique family of multifunctional proteins found in various tissues and cellular compartments. Initially, CLICs were considered to be solely involved in chloride... -


AffiDYE® GelRed (10,000X): Nucleic Acid Staining
Explore AffiDYE® GelRed, a groundbreaking nucleic acid stain that significantly enhances DNA and RNA visualization in laboratory settings. With its advanced formulation, AffiDYE® GelRed offers a safer, highly sensitive alternative to... -


AFFICHECK® Strep A PCR Controls
AffiCHECK®Strep A PCR Controls Positive Control Negative Control External Run Control Panel Control Verified Platforms Cepheid® GeneXpert® Luminex® Aries® AFG-CHK-0405 | AffiCHECK® Streptococcus A PCR... -


FeLV Feline Leukemia Virus
What is Feline Leukemia Virus ? Feline leukemia virus affected cats can develop anemia (a low red blood cell level), cancers, and/or suppression of the immune system. The disease worsens... -

AffiCHECK® PCR Controls by Equipment - Enhancing Diagnostic PCR Accuracy
Improve the accuracy of diagnostic PCR with AffiCHECK® PCR Controls by Equipment. Developed by AffiGEN®, a trusted provider of molecular biology solutions, these controls are designed to optimize PCR performance on various diagnostic equipment. -

AffiCHECK® PCR Controls by Pathogen
At AffiGEN®, we understand the critical role of PCR controls in ensuring the validity of your molecular biology experiments. AffiCHECK® PCR Controls specifically developed to validate and monitor PCR assays, allowing you to detect and amplify target sequences with utmost precision. Let's explore the classification of pathogens tested by PCR and the comprehensive coverage provided by AffiCHECK® PCR Controls. -


Quantify Human IL-1 beta Levels with High Precision using the Human IL-1 beta ELISA Kit
The Human IL-1 beta ELISA Kit is a powerful tool for researchers seeking to investigate the role of IL-1 beta in various physiological and pathological processes. This article provides a comprehensive overview of the assay protocol, highlighting the step-by-step instructions for sample preparation, plate setup, antibody incubation, washing, substrate addition, and data analysis. Furthermore, it delves into the wide range of applications where the Human IL-1 beta ELISA Kit can be employed, such as studying inflammation, immune responses, and related diseases. With its high sensitivity and specificity, this kit enables accurate quantification of IL-1 beta levels in human samples, providing valuable insights into the underlying mechanisms of various conditions. Discover the benefits of incorporating the Human IL-1 beta ELISA Kit into your research arsenal and unlock new avenues for understanding and targeting IL-1 beta-mediated pathways.





















